Mediterranean Anemia is a type of anemia that is usually seen in the Mediterranean region and can be passed on to subsequent generations through genetic factors. There are 1.4 million carriers and around 4500 thalassemia patients in our country.
Beta thalassemia, the most serious form of the disease, occurs after the baby turns 6 months old and can lead to organ damage. The disease is controlled with blood transfusions from the moment it is detected.
Today, stem cell transplantation is the definitive treatment for the disease. However, the fact that the organs are not damaged plays an important role in the success of the stem cell transplant.
People who are carriers of Mediterranean Anemia (Thalassemia) do not have any significant symptoms other than anemia and do not need treatment. Today, thanks to advances in medicine, thalassemia can be controlled by increasing the quality of life and life expectancy of patients.
How does Mediterranean Anemia occur?
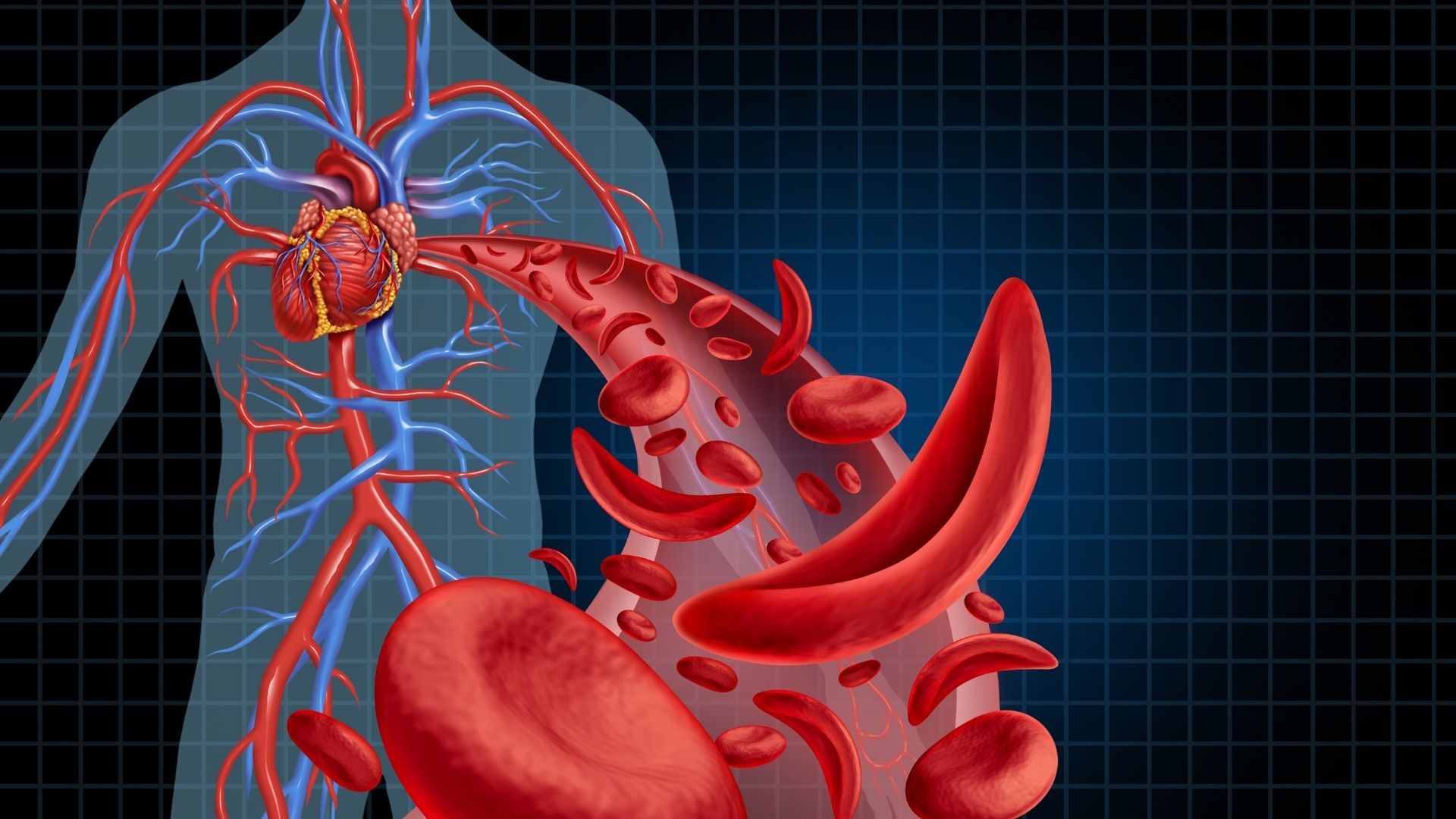
In thalassemia, there is a defect or deficiency in the genes involved in the production of hemoglobin, the substance that carries oxygen in red blood cells.
Hemoglobin in red blood cells in our blood carries the oxygen necessary for tissues. In case of insufficiency or defect in hemoglobin production, oxygen transport cannot be carried out sufficiently, and complaints such as pallor, weakness, fatigue, palpitations, developmental retardation develop as a result of decreased oxygen in tissues and organs.
What are the Causes of Mediterranean Anemia?
Mediterranean Anemia (Thalassemia) is caused solely by hereditary causes. One in every 40-50 people in Turkey and one in every 10 people in Antalya, Adana and the Southeastern Anatolia region carries the genes that can cause thalassemia. People have two genes for a trait; one is inherited from the mother and the other from the father. For Mediterranean anemia to occur, at least one of the two parents must be a carrier or have the disease. If the parents do not have the disease gene, the offspring will not have it either.
In case of carriage, the disease does not occur. It is important to inform people who are only carriers so that they can have healthy children in the future. Since the risk of thalassemia is especially high in consanguineous marriages, it is very important for these people to have the necessary tests done before marriage.
Symptoms and Treatment Methods
The symptoms of thalassemia vary depending on the severity. It usually starts to show symptoms a few months after birth. Mediterranean anemia can mimic iron deficiency due to symptoms such as pallor and fatigue. Therefore, it can be confused with anemia.
Patients are treated with regular blood transfusions. These transfusions are necessary for patients to survive. However, blood transfusions may cause iron accumulation. Therefore, it is important to monitor iron levels regularly and use iron excretion medications if necessary.
Bone marrow transplantation is currently accepted as the definitive treatment for thalassemia. It is performed by transferring stem cells from donors with appropriate tissue compatibility to the patient.
Mediterranean Anemia (Thalassemia) is a genetic disease and can be controlled with genetic counseling and appropriate treatment methods. It is of great importance for patients and carriers to follow the recommended steps in order to give birth to healthy children.